Trwa ładowanie...
Money.pl
Zaloguj
Giełda
Notowania GPW
Spółki GPW
Porównanie spółek
Giełda na żywo
Giełdy światowe
Komunikaty ESPI
Surowce
Narzędzia giełdowe
Portfel
Waluty
Kursy walut
Kalkulator walutowy
Bitcoin
Forex
Depozyty
Dla przedsiębiorców
Jak założyć firmę
Faktoring dla firm
Poradniki
PKD
PKWiU
Mobility
Praca
Oferty pracy
Wynagrodzenia
Kalkulator PPK
Kalkulator wynagrodzeń
Biznes i Gospodarka
BizTech
Ekologia, OZE, ESG
Nieruchomości i budownictwo
Emerytury
Prawo
Kariera i rynek pracy
Finanse osobiste
Kalkulatory
Kalkulator urlopu macierzyńskiego
Kalkulator odsetkowy
Kalkulator zakupów na firmę
Kalkulator składek ZUS
Kalkulator wynagrodzenia za pracę w nadgodzinach
Kalkulator dat
Kalkulator wynagrodzenia chorobowego
Kalkulator ekwiwalentu za urlop wypoczynkowy
Kalkulator stażu pracy
Kalkulator kredytu hipotecznego
Kalkulator składek ZUS dla działalności gospodarczej
Strefa money
Biznes Klasa
Program Money
Nagroda Money
Pomysł Na Dom
Stan gospodarki
Wybory parlamentarne 2023
Obietnice wyborcze 2023
Money Insights
Wydarzenia
Impact
Kongres 590
Forum Ekonomiczne
Szczyt Klimatyczny Togetair
Zapytaj Notariusza
Krynica Forum 2022
Krynica Forum 2023
Rynek nieruchomości - INPON 2023
Wyższy poziom chmury
Cybersec Forum
Kongres ESG Polska Moc Biznesu
II Europejski Kongres Sportu I Turystyki
Podatki
Wiadomości podatkowe
Pit
Formularze podatkowe
Poradniki podatkowe
Znajdź Adres
Prawo
Wiadomości
Twoje finanse (REKLAMA)
Fundusze
Wiadomości
Profile funduszy
Wyszukiwarka fundusz
Stopa zwrotu z funduszy
Kalkulator prowizji
Kalkulator zysku
Archiwum Funduszy
Banki
Poradniki
Ubezpieczenia
Emerytury
Poradniki
Giełda
NARZĘDZIA
Notowania GPW
Spółki GPW
Porównanie spółek
Giełda na żywo
Giełdy światowe
Komunikaty ESPI
Surowce
Narzędzia giełdowe
Portfel
Waluty
NARZĘDZIA
Kursy walut
Kalkulator walutowy
Bitcoin
Forex
Depozyty
Dla przedsiębiorców
Jak założyć firmę
Faktoring dla firm
Poradniki
NARZĘDZIA
PKD
PKWiU
Mobility
Praca
NARZĘDZIA
Oferty pracy
Wynagrodzenia
Kalkulator PPK
Kalkulator wynagrodzeń
Biznes i Gospodarka
BizTech
Ekologia, OZE, ESG
Nieruchomości i budownictwo
Emerytury
Prawo
Kariera i rynek pracy
Finanse osobiste
Kalkulatory
NARZĘDZIA
Kalkulator urlopu macierzyńskiego
Kalkulator odsetkowy
Kalkulator zakupów na firmę
Kalkulator składek ZUS
Kalkulator wynagrodzenia za pracę w nadgodzinach
Kalkulator dat
Kalkulator wynagrodzenia chorobowego
Kalkulator ekwiwalentu za urlop wypoczynkowy
Kalkulator stażu pracy
Kalkulator kredytu hipotecznego
Kalkulator składek ZUS dla działalności gospodarczej
Strefa
Biznes Klasa
Program Money
Nagroda Money
Pomysł Na Dom
Stan gospodarki
Wybory parlamentarne 2023
Obietnice wyborcze 2023
Money Insights
Wydarzenia
Impact
Kongres 590
Forum Ekonomiczne
Szczyt Klimatyczny Togetair
Zapytaj Notariusza
Krynica Forum 2022
Krynica Forum 2023
Rynek nieruchomości - INPON 2023
Wyższy poziom chmury
Cybersec Forum
Kongres ESG Polska Moc Biznesu
II Europejski Kongres Sportu I Turystyki
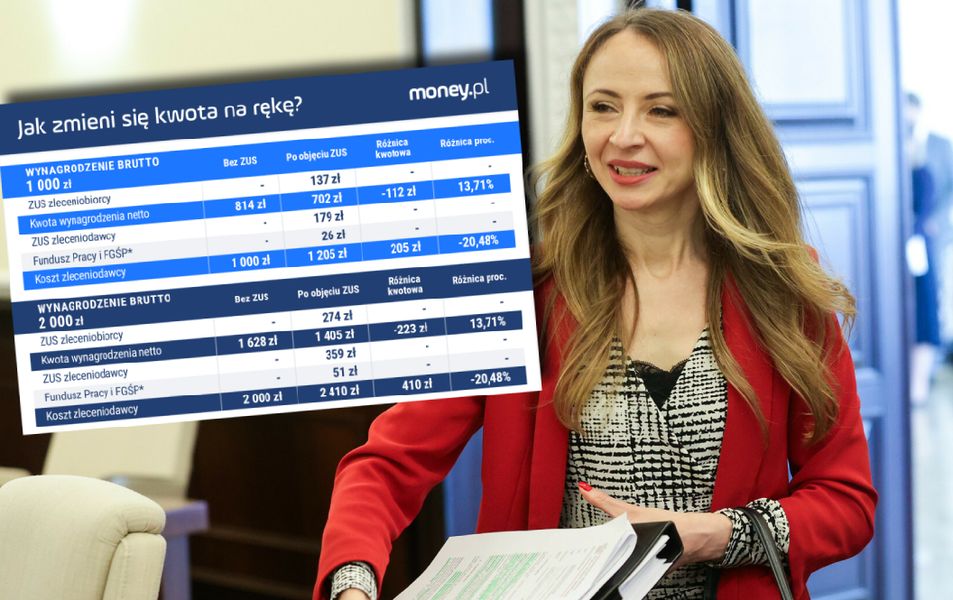
Małgorzata Samborska
Idzie wielka rewolucja w składkach. Oto jak wpłynie na portfele Polaków
Polska pod "żelazną kopułą". Niczego nie tracimy, a możemy tylko zyskać
Nowe zwolnienie z PIT. Decyzja w ostatniej chwili
Wielka wycinka pod elektrownię jądrową na Pomorzu. "Praca wre"
Kolejowe "szprychy" CPK do kosza? Burza w sieci. Mówią o sabotażu
Rząd nie spieszy się z decyzjami ws. energetyki. "To recepta na kryzys"
MAT. SPONSOROWANY
TOGETAIR 2024. Oglądaj na żywo. Scena CLIMATE CARE
Money Magazyn
TYLKO W MONEY
Białe skarpetki i klapki. Obciach i absurd obróciła w miliony złotych
Łukasz Kijek
TYLKO U NAS
"Biuro nieczynne". Firma upada, dziesiątki klientów na lodzie
Krystian Rosiński
"Zarabiałem 3,8 zł za godzinę". Dziś ma firmę z przychodem 200 mln zł
Łukasz Kijek
Gospodarka
Zmienili walutę na euro. Oto jak teraz wyglądają ceny
Marcin Walków
Sprzedaż detaliczna w Polsce. Są najnowsze dane
Ukraiński minister rolnictwa podejrzany o korupcję. Chodzi o przejmowanie ziemi
"To cud gospodarczy". Prezydent Argentyny chwali terapię szokową
Port Elbląg liczy na większy przeładunek. Szansa na rozwój
Ministrowie się porozumieli. Unia chce rozszerzyć sankcje na Iran
Problematyczny autobus linii 116. Barcelona uderza w turystów
Finanse
Dlaczego firmy uciekają z Polski? Eksperci wymieniają pięć powodów
Karolina Wysota
Polacy dostaną nowy bon. Co z podatkiem? Jest komunikat MF
Będzie uderzenie w portfele? Rząd "odkręca" reformę opłat za wodę i ścieki
Rozliczenie składki zdrowotnej. Ważny komunikat ZUS
Deweloperzy w blokach startowych. Szykują ofertę pod program i "liczą na powtórkę"
Lawinowy przyrost emerytur groszowych. "Trzeba wprowadzić staż minimalny"
Polacy coraz więcej wpłacają na internetowe zbiórki. Jak je rozliczyć?
Notowania
GIEŁDA
WALUTY
SUROWCE
Biznes
"Biuro nieczynne". Firma upada, dziesiątki klientów na lodzie
Artur Dziambor rzecznikiem przedsiębiorców? Premier przed decyzją
Na pozwolenie firmy czekają nawet 2 lata. Gigantyczny problem na budowach dróg
Ciech zmienia nazwę. Koncern polskiego miliardera ma nową strategię
Serwisowali samoloty w Jasionce. Zwolnili 60 osób. "Firma spakowała się"
Rynki
Giełda
Waluty
100 tys. dolarów za bitcoina? Eksperci: to możliwe jeszcze w tym roku
Co jest głównym silnikiem napędowym polskiej gospodarki? Te dane wszystko wyjaśniają
Są nowe dane zza Odry. Wiadomo co z gospodarką Niemiec
Jen na najniższym poziomie od 34 lat. Co się dzieje?
Unia Europejska wciąż ma problem z gospodarką? Są nowe dane
Wideo
Biznes Klasa
Pomysł na dom
Fala zwolnień w Polsce. Dominują te branże "Pęka bańka"
Michał Wąsowski
TYLKO W MONEY
Firmy zwalniają pracowników. "To są lokalne dramaty"
TYLKO W MONEY
Zaczynał na stadionie. Dziś ma 120 salonów i obraca milionami
Zarządza potężnym majątkiem polskiego miliardera. "Złoto się kończy"
Prawo
"Celowe dążenie do usunięcia prezesa". NIK zawiadamia prokuraturę
Paweł Gospodarczyk
Podwyższą zasiłek pogrzebowy. Policzyli, ile to będzie kosztować
Służby chcą rozszerzyć inwigilację. Ministerstwo pracuje nad ustawą
Centralne Biuro Zwalczania Korupcji zastąpi CBA. "Polska znacząco spadła w rankingach"
Energetyka
Polska może zamknąć jedną z procedur naruszeniowych UE. "Pracujemy nad tym"
Ojciec chrzestny fotowoltaiki przestrzega Europę. Wymienia jeden kraj
Ukraina wysłała prąd do Polski. Wszystko przez słoneczne dni
Praca i kariera
Zakład Levi Strauss do likwidacji. Jest porozumienie ws. odpraw
Rząd reaguje na zwolnienia grupowe. Sypnie pieniędzmi
Koniec znienawidzonej przez pracowników ery na rynku pracy. Prezeska firmy Kubota o wpływie pokolenia Z
Wybrane dla Ciebie
Najnowsze
Biznes
Finanse
Księgowość
Goesgreen
Zarobki